
Kevin M. Allan,
Boram D. Hong and
Brian M. Stoltz*
The Arnold and Mabel Beckman
Laboratories of Chemical Synthesis, Division of Chemistry and Chemical
Engineering, California Institute of Technology, 1200 E. California
Boulevard, MC 164-30, Pasadena, CA 91125, USA. E-mail: [email protected]; Fax: +1 626 564 9297; Tel: +1 626 395 6064
A convenient method is disclosed for the synthesis of both 3-hydroxyisoquinolines and 2-hydroxy-1,4-naphthoquinones from 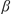 -ketoesters
using a one-pot aryne acyl-alkylation/condensation procedure. When
performed in conjunction with a one-step method for the synthesis of
the -ketoester
substrates, this method provides a new route to these polyaromatic
structures in only two steps from commercially available carboxylic
acid starting materials. The utility of this approach is demonstrated
in the synthesis of the atropisomeric P,N-ligand, QUINAP.
-ketoesters
using a one-pot aryne acyl-alkylation/condensation procedure. When
performed in conjunction with a one-step method for the synthesis of
the -ketoester
substrates, this method provides a new route to these polyaromatic
structures in only two steps from commercially available carboxylic
acid starting materials. The utility of this approach is demonstrated
in the synthesis of the atropisomeric P,N-ligand, QUINAP.
Synthetic chemists are constantly in search of new
reaction sequences to convert simple, readily available starting
materials into increasingly complex products. The most useful and
economically valuable of these processes are able to do so quickly and
with little operational difficulty.1 It was with this concept in mind that we designed a procedure for the conversion of -ketoesters
to either 3-hydroxyisoquinolines or 2-hydroxy-1,4-naphthoquinones using
a novel one-pot aryne acyl-alkylation/condensation sequence.
Furthermore, when this process is coupled with a one-step synthesis of
the -ketoester
substrates, it provides an exceptionally general two-step procedure for
the conversion of readily available carboxylic acids to either of these
important bicyclic aromatic structures.2
Traditional approaches to the incorporation of
these aromatic functionality within larger molecular scaffolds have
typically relied upon transition metal-catalyzed cross coupling
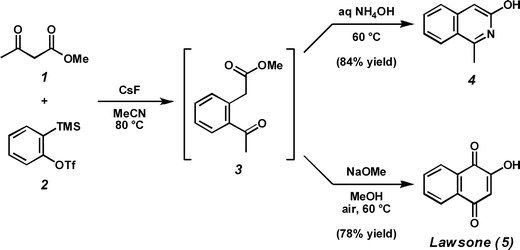
reactions employing C(sp2)–X precursors.3 As an alternative to this strategy, we have been exploring methods to prepare various heteroaromatic systems via condensation of arynes with derivatives of carboxylic acids.4 Following our report of a fluoride-induced insertion reaction between -ketoesters (1) and arynes derived from silyl aryl triflates (2),5 we began to investigate avenues by which the acyl-alkylated arene products (3) could be advanced toward larger ring systems (Scheme 1).6 Our interest in nitrogen-containing heterocycles7 led us to a report by Bentley et al., in which the synthesis of both 3-hydroxyisoquinolines (4) and 2-hydroxy-1,4-naphthoquinones (5) is accomplished by exposure of 1,5-ketoesters similar to 3 to either aqueous ammonia or alkaline base under an ambient atmosphere, respectively.8 Indeed, we found that treatment of a crude acyl-alkylation reaction mixture containing methyl (2-acetylphenyl)acetate (3) with aqueous ammonium hydroxide furnished 1-methyl-3-hydroxyisoquinoline (4),9 while addition of sodium methoxide generated lawsone (5), an isolate from the henna plant Lawsonia inermis.10
 |
||
| Scheme 1 Aryne acyl-alkylation followed by condensation with ammonia or intramolecular condensation and oxidation. | ||
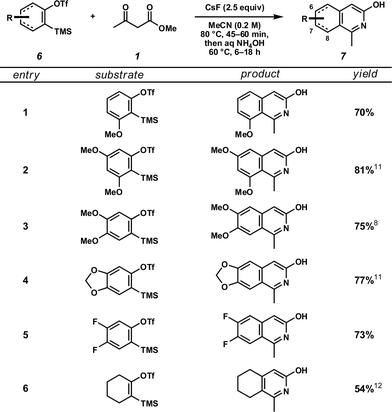
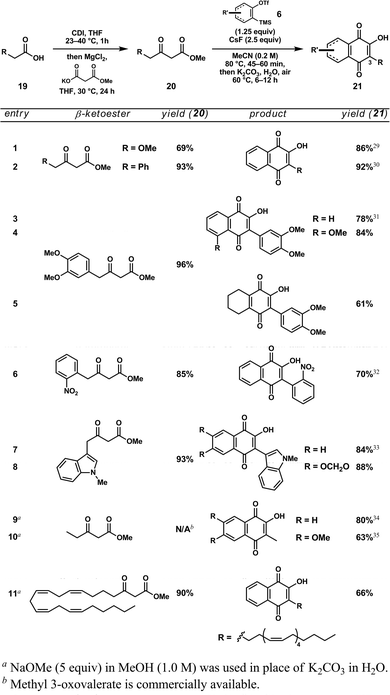
Focusing on the first of these two transformations, we investigated the reaction between methyl acetoacetate (1) and a number of functionalized aryne precursors (6) (Table 1). The sequence proved quite general, providing rapid access to a number of 3-hydroxyisoquinoline products (7)
featuring substitution at carbons 6, 7, and 8. The reaction proceeded
well with both electron-rich (entries 1–4) and electron-deficient
(entry 5) arynes, as well as with cyclohexyne13
(entry 6). Notably, the use of unsymmetrically substituted arynes
(entries 1 and 2) provided single product isomers originating from
regioselective addition of the -ketoester to the inductively activated carbon meta to the heteroatom.14
 |
Next, we considered alternative -ketoesters for the introduction of substitution at C(1) of the hydroxyisoquinoline scaffold (Table 2). Though methyl acetoacetate and a handful of other -ketoesters
are commercially available, we found it necessary to prepare additional
substrates in order to probe the scope of the reaction. While there are
a number of methods to generate -ketoesters from the corresponding carboxylic acids (8),
we selected a procedure reported by Masamune and coworkers, which
employs the addition of a magnesium methylmalonate to a preformed acyl
imidazole.15 Both simple and high yielding, this method facilitated the synthesis of a small library of differentially substituted methyl -ketoesters (9) from readily available materials in a single step.16
 |
With a collection of substrates in hand, we set
about testing the steric and electronic tolerances of our
acyl-alkylation/condensation reaction sequence. Under optimized
conditions, the -ketoester (9) and silyl aryl triflate (6) are combined in a Schlenk flask and heated in the presence of CsF. Following formation of the acyl-alkylated arene (e.g., 3),
ammonium hydroxide is added and the condensation is allowed to proceed
in a sealed environment. We were delighted to find that the reaction
proceeds quite well with a variety of alkyl (entries 1 and 2), aryl
(entries 3–5), and heteroaryl groups (entries 6–8) located at the 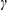 -position of the -ketoester. In addition, naphthyl isoquinoline (entry 9) and bis-isoquinoline (entry 10) scaffolds—structures central to many ligand frameworks—are accessible.18
Interestingly, this sequence proves quite effective at rapidly
appending isoquinoline motifs on to biological substrates such as
arachidonic acid (entry 11) and 3-acetoxy-5-etienic
acid (entry 12) without epimerization of existing stereocenters. The
reaction can even be performed bidirectionally with a diacid to
generate a bis(3-hydroxyisoquinoline)
product (entry 13). Furthermore, the hydroxyl group provides an
excellent functional handle for the introduction of additional
substitution at C(3) of the isoquinoline. As a demonstration of this
fact, we carried out the triflation of 3-hydroxyisoquinoline 11 (Table 2, entry 6) followed by a palladium-catalyzed Suzuki coupling with 4-methoxyphenylboronic acid (12) to furnish diarylisoquinoline 13 in high yield (Scheme 2).
Thus, this method provides avenues not only to C(1) functionalized
hydroxyisoquinolines, but toward 1,3-disubstituted isoquinolines as
well.
-position of the -ketoester. In addition, naphthyl isoquinoline (entry 9) and bis-isoquinoline (entry 10) scaffolds—structures central to many ligand frameworks—are accessible.18
Interestingly, this sequence proves quite effective at rapidly
appending isoquinoline motifs on to biological substrates such as
arachidonic acid (entry 11) and 3-acetoxy-5-etienic
acid (entry 12) without epimerization of existing stereocenters. The
reaction can even be performed bidirectionally with a diacid to
generate a bis(3-hydroxyisoquinoline)
product (entry 13). Furthermore, the hydroxyl group provides an
excellent functional handle for the introduction of additional
substitution at C(3) of the isoquinoline. As a demonstration of this
fact, we carried out the triflation of 3-hydroxyisoquinoline 11 (Table 2, entry 6) followed by a palladium-catalyzed Suzuki coupling with 4-methoxyphenylboronic acid (12) to furnish diarylisoquinoline 13 in high yield (Scheme 2).
Thus, this method provides avenues not only to C(1) functionalized
hydroxyisoquinolines, but toward 1,3-disubstituted isoquinolines as
well.
 |
||
| Scheme 2 Synthesis of a 1,3-diarylisoquinoline. | ||
As previously mentioned, our aryne
acyl-alkylation/condensation reaction sequence enables rapid assembly
of structural motifs that are key to several chiral ligand frameworks.
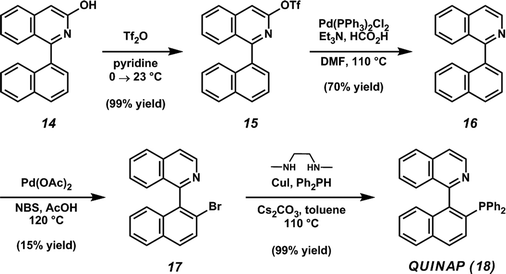
In particular, we envisioned that naphthyl isoquinoline 14 (Table 2, entry 9) could be employed toward the synthesis of 1-(2 -diphenylphosphino-1-naphthyl)-isoquinoline (QUINAP, 18),19
an axially chiral P,N-ligand that has found widespread application in
asymmetric catalysis, including enantioselective hydroboration,20 allylic alkylation,21 and hydrogenation22 (Scheme 3). Having developed an expedient method for the construction of naphthyl 3-hydroxyisoquinoline 14,
we set out to demonstrate the feasibility of our synthetic approach.
Following the acyl-alkylation/condensation sequence, the resulting
hydroxyisoquinoline (14) is subjected to triflation. Next, reduction of triflate 15 yields naphthyl isoquinoline 16, which is subsequently brominated via a nitrogen-directed palladium-catalyzed C–H activation.23 Finally, a modified version of Buchwald
-diphenylphosphino-1-naphthyl)-isoquinoline (QUINAP, 18),19
an axially chiral P,N-ligand that has found widespread application in
asymmetric catalysis, including enantioselective hydroboration,20 allylic alkylation,21 and hydrogenation22 (Scheme 3). Having developed an expedient method for the construction of naphthyl 3-hydroxyisoquinoline 14,
we set out to demonstrate the feasibility of our synthetic approach.
Following the acyl-alkylation/condensation sequence, the resulting
hydroxyisoquinoline (14) is subjected to triflation. Next, reduction of triflate 15 yields naphthyl isoquinoline 16, which is subsequently brominated via a nitrogen-directed palladium-catalyzed C–H activation.23 Finally, a modified version of Buchwald s phosphine–aryl bromide coupling24 affords the desired target, QUINAP (18).
Whereas the majority of previous approaches rely on transition
metal-catalyzed cross coupling reactions to form the axial C–C bond,25
our synthetic route represents a novel disconnection that ultimately
builds the isoquinoline ring system on to the existing framework of
naphthoic acid.
s phosphine–aryl bromide coupling24 affords the desired target, QUINAP (18).
Whereas the majority of previous approaches rely on transition
metal-catalyzed cross coupling reactions to form the axial C–C bond,25
our synthetic route represents a novel disconnection that ultimately
builds the isoquinoline ring system on to the existing framework of
naphthoic acid.
 |
||
| Scheme 3 Synthesis of QUINAP. | ||
The second class of compounds, 2-hydroxy-1,4-naphthoquinones, is generated via
intramolecular condensation of the acyl-alkylated arene to form an
intermediate 1,3-naphthalenediol, which then autooxidizes under an
ambient atmosphere. We first encountered these compounds while working
toward 3-hydroxyisoquinolines when we noticed that -ketoester substrates bearing an acidic -methylene (20) preferentially formed the hydroxynaphthoquinone, albeit in low yield, through the action of NH4OH
as a base. When aqueous potassium carbonate was added in its place and
the mixture was heated while open to air, the reaction proceeded
cleanly to the hydroxynaphthoquinone (21) (Table 3).26
Using these optimized conditions, we are able to generate a variety of
hydroxynaphthoquinones possessing alkoxy (entry 1), aryl (entries 2–6),
and even indole (entries 7 and 8) substitution at C(3). However, having
optimized the reaction for -ketoesters bearing electron-withdrawing functionality at the -position,
it was necessary to select a stronger base in order to target alkyl
substituted products. Fortunately, sodium methoxide provided a suitable
alternative in such cases, giving rise to simple natural product
scaffolds such as phthiocol27 (entry 9) and O-des-methyl-stoechadone28
(entry 10), as well as a derivative of arachidonic acid (entry 11).
Under both conditions, differentially substituted arynes (entries 4, 8,
and 10) and cyclohexyne (entry 5) performed comparably to benzyne
(derived from 2).
 |
In summary, we have developed an extremely concise
procedure for the synthesis of 3-hydroxyisoquinolines and
2-hydroxy-1,4-naphthoquinones from -ketoesters
using a novel one-pot aryne acyl-alkylation/condensation sequence. By
employing this procedure in conjunction with a one-step synthesis of -ketoesters
from carboxylic acids, we are able to build these bicyclic aromatic
structures in only two steps from commercially available materials.
Furthermore, this method is capable of joining a variety of
differentially functionalized -ketoester
and silyl aryl triflate substrates to generate products bearing a wide
range of steric and electronic substitution. The utility of this
approach has been demonstrated in the synthesis of the axially chiral
P,N-ligand, QUINAP. Given the proven scalability of the aryne
acyl-alkylation step,36
this approach is expected to enable the preparation of multigram
quantities of either of these product structures. Further investigation
of this methodology, including its utilization in more complex
settings, is currently underway in our laboratories and will be
reported in due course.
Representative procedure for the synthesis of 3-hydroxyisoquinolines from  -ketoesters
-ketoesters
A flame-dried 10 mL Schlenk flask with a septum-covered side arm equipped with a magnetic stir bar was charged with caesium fluoride (0.152 g, 1.00 mmol, 2.5 equiv). The flask was evacuated and back-filled with argon (×2). Acetonitrile (2 mL), methyl acetoacetate (1) (0.043 mL, 0.4 mmol, 1.0 equiv) and 2-(trimethylsilyl)phenyl trifluoromethanesulfonate (2) (0.121 mL, 0.498 mmol, 1.25 equiv) were sequentially added. The screw valve was sealed and the reaction was heated to 80 °C while stirring for 1 h. The reaction was cooled to room temperature when TLC analysis showed complete consumption of methyl acetoacetate (1). The screw valve was removed under positive argon pressure and aqueous ammonium hydroxide (28% w/w, 2 mL) was added via syringe. The screw valve was replaced and the reaction was heated to 60 °C while stirring for 8 h. The reaction was cooled to room temperature when TLC analysis showed complete consumption of the intermediate acyl-alkylated arene. The mixture was diluted with brine (5 mL) and extracted with EtOAc (2 × 15 mL). The aqueous layer was neutralized to pH 7 with 2.0 N HCl and extracted again with EtOAc (2 × 15 mL). The aqueous layers were discarded and the combined organic layers were extracted with 2.0 N HCl (3 × 20 mL). The organic layers were discarded and the combined aqueous layers were neutralized to pH 7 with 2.0 N NaOH and extracted with EtOAc (3 × 20 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified via recrystallization from boiling EtOAc to yield 1-methyl-3-hydroxyisoquinoline (4) as a yellow solid (0.0533 g, 84% yield).37
Representative procedure for the synthesis of 2-hydroxy-1,4-naphthoquinones from -ketoesters
A flame-dried 15 mL reaction tube equipped with a magnetic stir bar was charged with caesium fluoride (0.152 g, 1.00 mmol, 2.5 equiv). The reaction tube was sealed with a rubber septum, evacuated, and back-filled with argon (×2). Acetonitrile (2 mL), methyl 3-oxo-4-phenylbutanoate (Table 3, entry 2) (0.077 g, 0.4 mmol, 1.0 equiv) and 2-(trimethylsilyl)phenyl trifluoromethanesulfonate (2) (0.121 mL, 0.498 mmol, 1.25 equiv) were sequentially added. The reaction was then heated to 80 °C while stirring for 1 h. The reaction was cooled to room temperature when TLC analysis showed complete consumption of methyl 3-oxo-4-phenylbutanoate. Potassium carbonate (0.276 g, 5.0 equiv) in water (2 mL) was added via syringe and the biphasic mixture was vigorously stirred at room temperature for 30 min. The septum was then removed and the reaction was heated to 60 °C while open to air for 12 h. The reaction was cooled to room temperature when TLC analysis showed complete consumption of the acyl-alkylated arene intermediate. The reaction was diluted with EtOAc (10 mL) and extracted with 1.0 N K2CO3 (5 × 15 mL). The organic layer was discarded. The combined aqueous layers were acidified to pH 1 with 2.0 N HCl and extracted with EtOAc (3 × 40 mL). The combined organic layers were washed with brine (50 mL) and dried over MgSO4. After filtration, the solvent was removed under reduced pressure and the residue was purified via flash chromatography (SiO2, 10:90 EtOAc/hexanes) to yield 2-hydroxy-3-phenyl-1,4-naphthoquinone as a yellow solid (0.0918 g, 92% yield).37
The authors gratefully acknowledge Pamela M. Tadross and Scott C. Virgil for helpful discussions and experimental assistance. The authors thank Abbott, Amgen, Boehringer-Ingelheim, Bristol-Myers Squibb, Merck, Sigma-Aldrich and Caltech for generous funding.
| 1 | (a) The Way of Synthesis, ed. T. Hudlicky, and J. W. Reed, Wiley-VCH, Weinheim, 2007 |
|
| 2 | (a) Vitamins and Hormones, ed. G. Litwack, Academic Press, London, 2008, vol. 78. |
|
| 3 | (a) A. Krasovskiy, V. Krasovskaya and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 2958–2961 [Links]; (b) A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 [Links]; (c) S. C. Tucker, J. M. Brown, J. Oakes and D. Thornthwaite, Tetrahedron, 2001, 57, 2545–2554 [Links]; (d) J.-M. Valk, T. D. W. Claridge, J. M. Brown, D. Hibbs and M. B. Hursthouse, Tetrahedron: Asymmetry, 1995, 6, 2597–2610 [Links]; (e) T. R. Kelly, A. Garcia, F. Lang, J. J. Walsh, K. V. Bhaskar, M. R. Boyd, R. Gotz, P. A. Keller, R. Walker and G. Bringmann, Tetrahedron Lett., 1994, 35, 7621–7624 [Links]; (f) K. Tamao, S. Kodama, I. Nakajima and M. Kumada, Tetrahedron, 1982, 38, 3347–3354 [Links]. | |
| 4 | (a) K. M. Allan and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 17270–17271 |
|
| 5 | U. K. Tambar and B. M. Stoltz, J. Am. Chem. Soc., 2005, 127, 5340–5341 [Links]. | |
| 6 | U. K. Tambar, D. C. Ebner and B. M. Stoltz, J. Am. Chem. Soc., 2006, 128, 11752–11753 [Links]. | |
| 7 | (a) S. Krishnan, J. T. Bagdanoff, D. C. Ebner, Y. K. Ramtohul, U. K. Tambar and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 13745–13754 [Links]; (b) S. Krishnan and B. M. Stoltz, Tetrahedron Lett., 2007, 48, 7571–7573 |
|
| 8 | H. R. Bentley, W. Dawson and F. S. Spring, J. Chem. Soc., 1952, 1763–1768 [Links]. | |
| 9 | For the first preparation of this compound, see: B. Alpha, E. Anklam, R. Deschenaux, J.-M. Lehn and M. Pietraskiewiez, Helv. Chim. Acta, 1988, 71, 1042–1052 [Links]. | |
| 10 | G. Tommasi, Gazz. Chim. Ital., 1920, 50, 263 |
|
| 11 | For the first preparation of this compound, see: R. M. Kanojia, J. B. Press, O. W. Lever, Jr., L. Williams, J. J. McNally, A. J. Tobia, R. Falotico and J. B. Moore, Jr., J. Med. Chem., 1988, 31, 1363–1368 [Links]. | |
| 12 | For the first preparation of this compound, see: F. Freeman, D. K. Farquhar and R. L. Walker, J. Org. Chem., 1968, 33, 3648–3650 |
|
| 13 | (a) B. Iglesias, D. Peña, D. Pérez, E. Guitián and L. Castedo, Synlett, 2002, 486–488 |
|
| 14 | (a) E. R. Biehl, A. Razzuk, M. V. Jovanovic and S. P. Khanapure, J. Org. Chem., 1986, 51, 5157–5160 |
|
| 15 | D. E. Brooks, L. D.-L. Lu and S. Masamune, Angew. Chem., Int. Ed. Engl., 1979, 18, 72–74 [Links]. | |
| 16 | All carboxylic acid starting materials employed in Tables 2 and 3 are commercially available. | |
| 17 | For the first preparation of this compound, see: R. Nowicki and A. Fabrycy, Chem. Heterocycl. Compd., 1976, 12, 910–914 |
|
| 18 | (a) S. Busato, D. C. Craig, Z. M. A. Judeh and R. W. Read, Tetrahedron, 2003, 59, 461–472 [Links]; (b) M. B. Andrus and B. B. V. S. Sekhar, J. Heterocycl. Chem., 2001, 38, 1265–1271 [Links]; (c) G. Chelucci, A. Bacchi, D. Fabbri, A. Saba and F. Ulgheri, Tetrahedron Lett., 1999, 40, 553–556 [Links]. | |
| 19 | N. W. Alcock, J. M. Brown and D. I. Hulmes, Tetrahedron: Asymmetry, 1993, 4, 743–756 [Links]. | |
| 20 | (a) J. M. Brown, D. I. Hulmes and T. P. Layzell, J. Chem. Soc., Chem. Commun., 1993, 1673–1674 [Links]; (b) E. Fernandez, K. Maeda, M. W. Hooper and J. M. Brown, Chem.–Eur. J., 2000, 6, 1840–1846 [Links]. | |
| 21 | J. M. Brown, D. I. Hulmes and P. J. Guiry, Tetrahedron, 1994, 50, 4493–4506 [Links]. | |
| 22 | X. Li, L. Kong, Y. Gao and X. Wang, Tetrahedron Lett., 2007, 48, 3915–3917 [Links]. | |
| 23 | (a) D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Tetrahedron, 2006, 62, 11483–11498 [Links]; (b) D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Org. Lett., 2006, 8, 2523–2526 [Links]; (c) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300–2301 [Links]. | |
| 24 | K. Tani, D. C. Behenna, R. M. McFadden and B. M. Stoltz, Org. Lett., 2007, 9, 2529–2531 |
|
| 25 | (a) T. Thaler, F. Geittner and P. Knochel, Synlett, 2007, 17, 2655–2658 |
|
| 26 | Bentley et al. made a similar observation using methyl 2-(4,5-dimethoxy-2-acetylphenyl)acetate and later optimized the reaction for the formation of the hydroxynaphthoquinone in 53% yield using aqueous sodium hydroxide. See ref. 8. | |
| 27 | R. J. Anderson and M. S. Newman, J. Biol. Chem., 1933, 101, 773–779 |
|
| 28 | A. M. Aguinaldo, J. A. Armstrong, J. R. Cannon, S. M. Colegate, M. F. Comber, N. G. Marchant and M. V. Sargent, Aust. J. Chem., 1996, 49, 197–198 |
|
| 29 | For the first preparation of this compound, see: R. G. Cooke and W. R. Owen, Aust. J. Chem., 1962, 15, 486–491 |
|
| 30 | For the first preparation of this compound, see: T. Zincke and A. Breuer, Justus Liebigs Ann. Chem., 1884, 226, 22–60 |
|
| 31 | For the first preparation of this compound, see: G. Wurm and H.-J. Gurka, Pharmazie, 1997, 10, 739–743 |
|
| 32 | For the first preparation of this compound, see: K. Kobayashi, T. Taki, M. Kawakita, M. Uchida, O. Morikawa and H. Konishi, Heterocycles, 1999, 51, 349–354 |
|
| 33 | For the first preparation of this compound, see: S. Koulouri, E. Malamidou-Xenikaki, S. Spyroudis and M. Tsanakopoulou, J. Org. Chem., 2005, 70, 8780–8784 |
|
| 34 | For the first preparation of this compound, see: R. J. Anderson and M. S. Newman, J. Biol. Chem., 1933, 103, 405–412 |
|
| 35 | For the first preparation of this compound, see: A. C. Baillie and R. H. Thomson, J. Chem. Soc. C, 1966, 2184–2186 [Links]. | |
| 36 | D. C. Ebner, U. K. Tambar and B. M. Stoltz, Org. Synth., 2009, 86, 161–171 |
|
| 37 | See ESI for detailed spectroscopic data and experimental spectra. |
Footnote |
 Electronic supplementary information (ESI) available: General
experimental procedures, characterization data, NMR, and IR spectra.
See DOI: 10.1039/b913336d
Electronic supplementary information (ESI) available: General
experimental procedures, characterization data, NMR, and IR spectra.
See DOI: 10.1039/b913336d |
|
|
| This journal is © The Royal Society of Chemistry 2009 |